Seqboot und Consense
Seqboot 
Hiermit möchte ich ihnen eine kurze Einführung in die Funktionsweise und den Nutzen von Seqboot geben.
Seqboot ist ein Programm, dass geschrieben wurde, um an biologischen Datensätzen anhand von 3 Methoden Veränderungen durchzuführen.
Hierbei werden, aus einem bereits vorhandenen Datensatz beliebig viele abweichende Datensätze erzeugt, welche man dann getrennt analysieren kann. Auch wenn die Bootstrappingmethode schon 1979 von Bradley Efron erfunden wurde kam sie erst 1985 in der genetischen Analyse zum Einsatz. Das hier vorgestellte Programm Seqboot wurde 1985 von Felsenstein entwickelt. Es wird mit dem ebenfalls von Felsenstein entwickelten Programm Consense in der ebenfalls von ihm veröffentlichten Programmsammlung Phylib angeboten.
Seqboot beinhaltet drei verschiedene Methoden, um die Datensätze zu verändern:
1. Bootstrapping: Hierbei werden an dem bestehenden Datensatz entweder Lücken eingefügt, oder Teile vervielfältigt, wobei andere Teile überschrieben werden.
2. Jackkniffing: Hierbei wird willkürlich öfter die Hälfte des Datensatzes entfernt, so dass nur die anderen Hälften verglichen werden können.
3. Permuting: Verändert die Reihenfolge der Daten willkürlich und überprüft, ob dies zu weniger Gemeinsamkeiten führt. Ist dies der Fall kann von keiner Verwandschaft ausgegangen werden.
Nun möchte ich etwas genauer auf das Bootstrapping eingehen, da es die, in der Genetik, am meisten genutze Methode ist.
Bootstrapping ist eine Methode, um Datensätze zu verändern, ohne ihre Größe zu ändern. Dies geschieht durch das verdoppeln von einzelnen Werten oder das ersetzen durch Lücken. Die hierdurch veränderten Datensätze weichen also nur an manchen Stellen von dem eingegebenden Datensatz ab. Diese Veränderungen sollen die natürliche Varianz unter verschiedenen Stichproben simulieren.
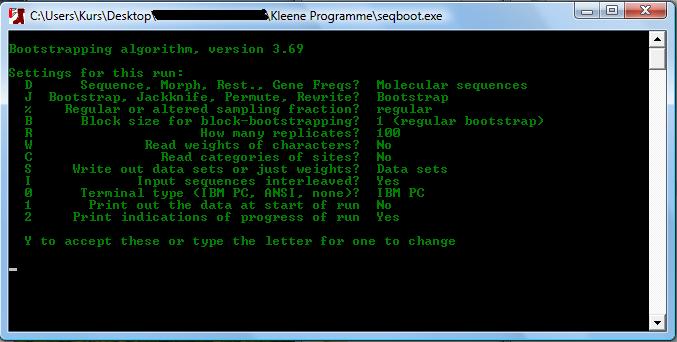 |
| Abb.1.: Hier sieht man Seqboot mal ausgeführt |
Wie zu sehen ist gibt es mehrere verschiedene Optionen, die man verändern kann. Da es jedoch sehr ausführlich wäre sie hier alle zu erklären. Verweise ich hierzu auf die Dokumentation des Programmes.
Seqboot ist sehr nützlich, wenn man z.B. überprüfen möchte, ob ein Stammbaum, der von einem anderen Programm erstellt wird, statistisch relevant ist.
Hierzu erstellt man mit Seqboot mehrere ähnliche Datensätze, aus welchen man dann Stammbäume erstellt. Wenn nun ein Großteil der minimal veränderten Stammbäume die selbe Taxonomie aufweist, stützt dies die Signifikanz dieser Taxonomie.
Da der manuelle Vergleich von so vielen Stammbäumen jedoch sehr mühsam sein kann enthält Phylib noch ein weiteres Programm, welches diese Aufgabe deutlich erleichtert. Es nennt sich Consense.
Consense 
Wie bereits erwähnt wurde Consense auch von Felsenstein entwickelt und wird in der von ihm entwickelten Programmsammlung Phylib angeboten. Consense ist ein Programm, welches in der Lage ist aus vielen verschiedenen variierenden taxonomischen Bäumen den mit der größten Häufigkeit zu finden. Also findet es den Baum, der von den meisten einzelnen Bäumen gestützt wird und gibt zu jedem Ast auch noch die Anzahl, der ihn stützenden Einzelbäume an.
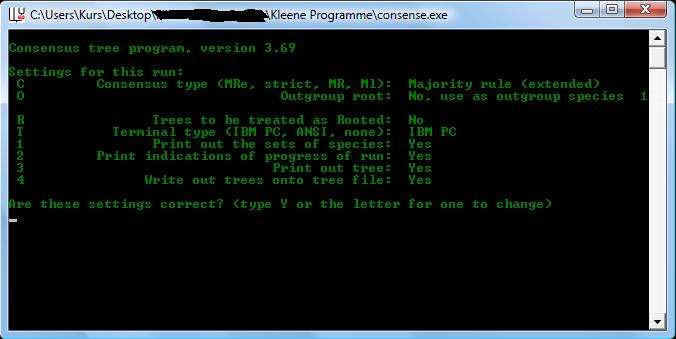 |
| Abb.2.: Hier sieht man Consense ausgeführt |
Da es auch hier viele verschiedene Optionen gibt, verweise ich wieder auf die ausführliche Dokumentation.
Als Beispiel habe ich hier nun mal einen von Consense erstellten Stammbaum.
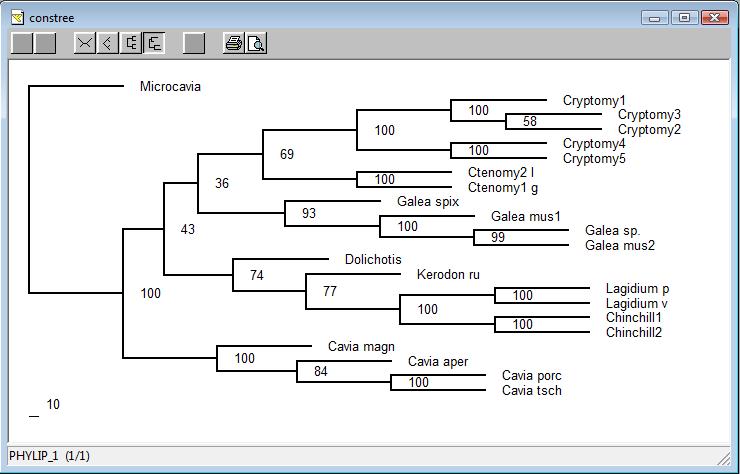 |
| Abb.3.: Von Consense erstellter Stammbaum (in Treeview dargestellt). |
Die jeweils an den Aufspaltungen vorhandenen Zahlen geben die Anzahl der geseqbooteten Bäume an, die diese Aufspaltung aufwiesen. Dies bezieht sich immer auf die Anzahl der Datensätze (in diesem Fall 100).
Falls sie das ganze also mal ausprobieren möchten, dann besuchen sie doch die Seite von Phylib (Link: http://evolution.gs.washington.edu/phylip/) und downloaden sie sich das gesamte Paket.