Genvorhersage/Gen-Annotation
Unter diesen Begriffen versteht man das a priori Auffinden von Genen innerhalb einer Nukleotidsequenz anhand von typischen Mustern wie beispielsweise Promotor, oder Start- und Stopsignale von Introns.

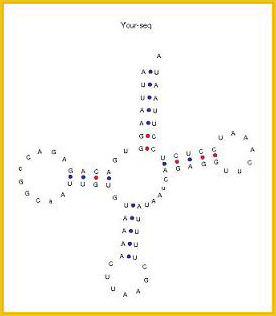
Das Programm identifiziert in gegebenen Nukleotidsequenzen Bereiche, die für Transfer-RNAs (tRNAs) codieren. Die aktuelle Version 1.23 wurde von Todd Lowe und Sean Eddy (University of California, Santa Cruz/Washington University, St. Louis) 2002 in PERL geschrieben. Das Programm kombiniert drei unabhängige Programme zur Vorhersage vont RNAs: tRNA-scan 1.3, einen von Pavesi et. al. entwickelter Algorithmus in der Implementierung Eufindtrna, und das Programm Cove, wobei die ersten zwei Programme zum Vorfiltern dienen, und Cove anschließend die Kandidaten-tRNAs mittels eines hochselektiven Covararanzmodells analysiert. Daten werden als RNA oder DNA Sequenz im fasta-Format hochgeladen. Das Ergebnis wird in Form einer Tabelle, einer Sekundärstrukur (s. Abbildung) oder im AceBD-Format (ein Datenbank-Format) ausgegeben.
ARWEN wurde entwickelt um tRNAs speziell in mitochondrialen Genomen (bei Metazoa) aufzuspüren. Die Motivation für das Programm lag darin begründet, dass gängige Programm zur Vorhersage von tRNAs bei mitochondrialen Genomen nur unzufriedenstellende Ergebnisse liefern - u. a. deswegen, weil die tRNAs der Mitochondrien im Vergleich zu cytosolischen und prokaryotischen tRNAs häufig degeneriert sind - es fehlen z. B. ganze Schleifen - und ausserdem weil Mitochondrien oft einen genetischen Code verwenden, der vom universellen Code abweicht (Basen wie Inosin, Pseudouridin, unterschiedlich methylierte Basen).
Die zu untersuchende Sequenz wird als fasta-Datei hochgeladen, als Ergebnis erhält man die Sekundärstrukturen der gefundenen tRNAs mit einigen Angaben wie Anzahl der Nukleotide, GC-Gehalt, und korrespondierende Aminosäure. Author des Programms ist Dean Laslett, die aktuelle Version 1.2 ist vom Jahr 2008.
Eine Gegenüberstellung der Analyseergebnisse von ARWEN und tRNA-scan SE kann z. B. folgendes Bild liefern:
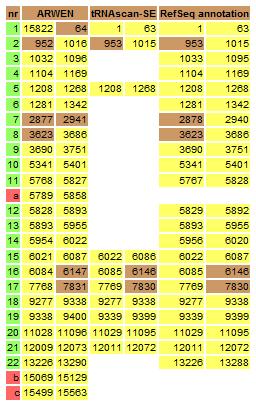
Es wurde das mitochondriale Genom von Artemia franciscana untersucht; die Referenzsequenz-Annotation stammt aus dem Vergleich mit einem nah verwandten Organismus. Die rot unterlegten Buchstaben kennzeichnen falsch annotierte tRNAs. Es wird die Überlegenheit von ARWEN gegenüber trna-scan SE bei Mitochondrien- tRNAs deutlich; wenn auch bei einigen Organismen der Vergleich zu Gunsten für tRNA-scan ausfällt. Die hoheSelektivität von ARWEN führt zwar zu einer fast 100%igen Erkennung aller tRNAs, hat aber auch den Nachteil, dass zusätzliche, falsch annotierte tRNAs angegeben werden. Da dies bei tRNA-scan SE fast nie vorkommt (dafür werden weniger tRNAs erkannt), führt die Kombination beider Verfahren zu sehr zuverlässigen Ergebnissen bei der tRNA-Annotation mitochondrialer Metazoa-Genome.
Literatur:
Laslett D, Canback B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24:172-175, PMID: 18033792